
Fibrosis Quística
Es una enfermedad crónica y hereditaria.
Está causada por un gen defectuoso que produce una proteína alterada implicada en el transporte de cloro. Esta proteína se encuentra en la membrana de muchas células epiteliales y glándulas exocrinas. Como consecuencia de esto, se producen problemas en:
- el aparato respiratorio
- el aparato genital masculino
- el páncreas
- el sistema hepatobiliar
- las glándulas sudoríparas
Es una enfermedad autosómica recesiva, es decir, se transmite de padres a hijos cuando el hijo hereda un gen mutado o defectuoso de cada uno de los padres. Es más frecuente en la raza caucásica.
Desde 2015, a todo niño recién nacido se le realiza la prueba del talón para determinar si tiene o no esta enfermedad.
El diagnóstico temprano es muy importante para poder alargar la esperanza de vida, así como para mejorar la calidad de ésta.
¿Cuáles son las causas de su aparición?
Se produce por la mutación del gen CFTR localizado en el cromosoma 7. Este gen produce la proteína reguladora de la conductancia transmembrana (CFTR). Esta proteína se encuentra en muchas células epiteliales y tiene diversas funciones:
- Forma un canal de cloro à implicado en el transporte de los iones de cloro.
- Regulación del flujo de otras sustancias à p.ej. sodio y agua.
- Regulación de otros canales de secreción celular à p.ej. en el páncreas consigue una concentración elevada de bicarbonato.
- Regulación de otros canales iónicos
La producción de la proteína CFTR alterada puede poner de manifiesto problemas con distinta gravedad y pronóstico. Se pueden producir distintas mutaciones o alteraciones que afectarán a las funciones anteriormente descritas. Dependiendo de si estas funciones se ven afectadas de forma total o parcial, la enfermedad se presentará de forma más o menos grave.
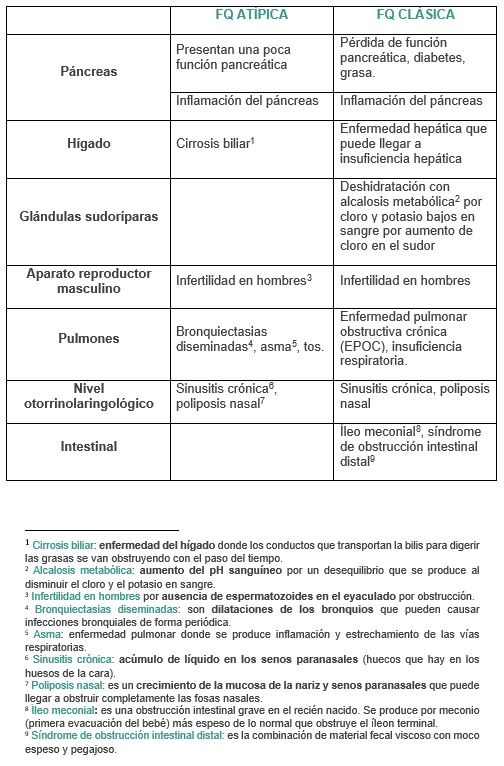
Según la afección del paciente, la fibrosis quística (FQ) se puede clasificar en FQ atípica y FQ clásica.
FQ ATÍPICA
- Presentan disfunción de la proteína CFTR debido a mutaciones en uno de los 2 cromosomas y además son mutaciones leves.
- Solamente se ve afectado un órgano.
- Puede llevar una vida normal.
- Los síntomas suelen aparecer a partir de los 10 años.
- Presenta mayor esperanza de vida que la FQ clásica.
FQ CLÁSICA
- Presentan mutaciones graves en al menos uno de los 2 cromosomas.
- Presentan prueba de sudor patológica o positiva.
- Presentan graves complicaciones.
- Los síntomas empiezan desde el nacimiento. Se dan desde los descritos en la FQ atípica hasta los descritos en la FQ clásica.
¿Qué síntomas son los más frecuentes?
Las manifestaciones clínicas dependen de la afectación de cada persona. En todos los afectados se producirá una progresión de la enfermedad a lo largo de su vida.
Tipos de tratamientos y algunos ejemplos.
- Tratamiento antibiótico: para disminuir el número de gérmenes que causan inflamación de las vías respiratorias. Se pueden administrar vía intravenosa, oral o inhalada, dependiendo de la gravedad de la infección.
- Broncodilatadores
- Antiinflamatorios: ibuprofeno y corticoides.
- Suplementos enzimáticos:
- Extractos pancreáticos (lipasa, amilasa y proteasa): facilitan la digestión cuando hay insuficiencia pancreática.
- Vitaminas liposolubles (A y E)
- Vitamina K
- Dornasa alfa inhalada: disminuye la viscosidad del moco presente en las secreciones bronquiales.
- Soluciones salinas hipertónicas inhaladas: para mejorar la función pulmonar.
- Fisioterapia respiratoria: para limpiar las secreciones espesas presentes en las vías respiratorias.
- Dispositivos mecánicos (como el flutter)
- Ejercicio físico
- Percusión de tórax
- Técnicas específicas de respiración
- Tos
- Tratamiento nutricional: seguir una dieta que mantenga un estado nutricional adecuado mejora la calidad de vida y la supervivencia.
- Trasplante pulmonar: última opción terapéutica tras fracaso de otras terapias.
- Nuevas terapias: hay fármacos potenciadores y correctores de la actividad de la proteína CFTR, por lo que se usan en combinación.
- POTENCIADORES:
- Ivacaftor: aumenta el transporte de cloro.
- CORRECTORES: mejoran el procesamiento y el transporte celular de CFTR, aumentando la cantidad de CFTR funcional en las células.
- Lumacaftor
- Tezacaftor
- Elexacaftor
- POTENCIADORES:
- Terapia intracelular: en investigación
- Terapia génica: en investigación.
El tratamiento debe individualizarse en cada caso, según la afección y gravedad de la enfermedad.
Consideraciones en población pediátrica y adolescente
Toda la información contenida en esta ficha es aplicable a la población pediátrica y adolescente.
Los síntomas de la FQ pueden aparecer al poco tiempo de nacer. En los recién nacidos, el primer síntoma suele ser una obstrucción intestinal que se conoce como “Ileo meconial”. La mayoría de los niños reciben el diagnóstico antes de los 2 años de edad al presentar infecciones pulmonares con aumento de moco, diarrea y retraso en el crecimiento. Sin embargo, algunos pueden tener síntomas más leves y no ser diagnosticados hasta la adolescencia.
La alimentación es un pilar fundamental en el control de la enfermedad. Aún tomando suplementos de enzimas pancreáticas, muchos niños tienen dificultades para absorber los nutrientes correctamente, por lo que en ocasiones necesitan seguir una dieta rica en alimentos calóricos y grasas que les permita mantener un peso y un crecimiento adecuados. Consulte a su médico o profesional de la nutrición si su hijo necesita seguir una dieta personalizada para cubrir sus necesidades nutricionales.
Los niños con FQ pierden más sal por el sudor que otros niños de su edad. Es recomendable conocer los signos de deshidratación como decaimiento, mareos o dolor de cabeza y tener a disposición suplementos de sal, agua o alimentos salados en días de calor excesivo o cuando se realiza ejercicio físico intenso.
Los niños con FQ pueden, y deben, hacer todo tipo de actividades, salir al patio con normalidad y ser uno más, tanto en el ámbito escolar como en el extraescolar. En cuanto al ejercicio físico y al deporte, se ha demostrado que mejora la tolerancia al ejercicio, mejora la capacidad pulmonar y aumenta la fuerza muscular y la masa ósea. El mejor deporte es el que guste a cada niño, ya que lo importante es que lo practique y sea constante. Sin embargo, hay deportes que conllevan más riesgos, como pueden ser aquellos realizados en altura, bajo el agua o en ambientes húmedos y con altas temperaturas.
Generalmente, los niños con FQ requieren tomar varios medicamentos al día, algo que puede ser un reto, especialmente para los adolescentes, que empiezan a ser más autónomos con sus tratamientos y cuidados. Por ello, es importante crear rutinas, tanto en casa como fuera de casa, que faciliten la toma de medicación diaria y educar en la importancia de cumplir con el tratamiento para un adecuado control de la enfermedad y un mejor pronóstico de la misma.
El plan de tratamiento ha de ser coordinado y conocido por todas las personas que rodean al niño, por ello, es recomendable facilitar información e instrucciones de actuación a profesores y personal que esté a cargo de su hijo en horario escolar.



Fecha de actualización:
08/12/2024
Autoría y revisión:
NEUMOLOGIA
Coautores:
GEFP